
 W4M : http://workflow4metabolomics.org/
W4M : http://workflow4metabolomics.org/
Workflow4Metabolomics is a collaborative portal dedicated to the processing, analysis and annotation of metabolomics data.The French Bioinformatics Institute (IFB) and MetaboHUB developed the full pipelines of LC/MS, GC/MS and NMR using the Galaxy framework for the analysis of data, including preprocessing, standardization, quality control, statistical analysis (univariate, multivariate PLS/OPLS) and annotation steps.
 PeakForest : https://peakforest.org/
PeakForest : https://peakforest.org/
The MetaboHUB PeakForest database provides storage and annotation services for metabolic profiles of biological matrices and reference. Through its Web portal, PeakForest is devoted to the annotation broadband and identification of de novo metabolites. It is based on the wide range of complementary methods using UPLC- (API) HRMS, the GC-QToF and NMR. This database is available in the MetaboHUB platforms to perform non-targeted metabolomic analyses of biofluids (eg, human plasma and urine), tissue samples (for example, the tomato fruit) and cell extracts (e.g., E. coli and S. cerevisiae). NMRProcFlow: http://www.nmrprocflow.org/
NMRProcFlow: http://www.nmrprocflow.org/
NMRProcFlow free software provides a complete set of tools for processing and visualization of 1D NMR data, all within an interactive interface based on a viewing of the spectra. BiotStatFlow : http://biostatflow.org/
BiotStatFlow : http://biostatflow.org/
BioStatFlow facilitates access to statistical tools for biologists who are not specialists. It has been designed to perform statistical analyses sequentially, namely a linear chain of statistical analysis, so-called Workflow for the "omic" data. MetExplore : http://metexplore.toulouse.inra.fr/joomla3/
MetExplore : http://metexplore.toulouse.inra.fr/joomla3/
MetExplore is a free service that allows academic:
- import, storage and sharing of metabolic networks across the genome
- mapping poly-omics data
- enrichment of metabolic pathways
- viewing networks
- extraction of networks from data and the network structure
- IT flux
MS-CleanR: A package for cleaning and annotating LC-MS data
The MS-CleanR package provides functions for feature filtering and annotation of LC-MS data.
See the publication and tutorials (pdf files included in the master branch) for more information.
Needs MS-DIAL (v4.00 or higher) and MS-FINDER (3.30 or higher): http://prime.psc.riken.jp/compms/index.html
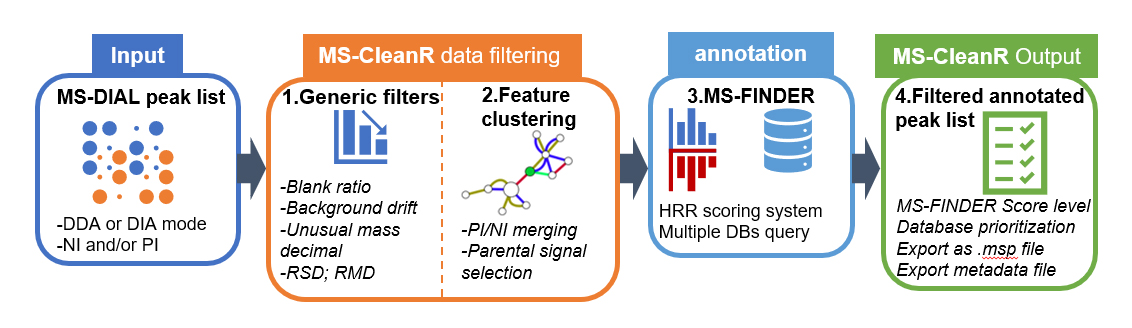
MS-CleanR use as input MS-DIAL peak list processed in data dependent analysis (DDA) or data independent analysis (DIA) using either positive ionization mode (PI) or negative ionization mode (NI) or both. First, MS-CleanR apply generic filters encompassing blank injection signal subtraction, background ions drift removal, unusual mass defect filtering, relative standard deviation threshold (RSD) based on sample class and relative mass defect (RMD) window filtering. All these options are tunable by the user. The second step involves a feature clustering method based on MS-DIAL peak character estimation algorithm followed by parental signal extraction using multi-level optimization of modularity algorithm. Optionally, MS-CleanR can merge PI and NI mode during this step. Then, all selected features are exported to MS-FINDER program for in silico-based annotation using hydrogen rearrangement rules (HRR) scoring system. At this step, multiple databases can be queried and each annotation results will be handled by MS-CleanR. The final step will merge annotation results to the filtered peak list by prioritizing database annotation depending on user choice. Optionally, all results can be exported as .msp file for mass spectral similarity networking purpose.
Installation
devtools::install_github("eMetaboHUB/MS-CleanR") library(mscleanr) runGUI()
Citation
Publication link: https://www.biorxiv.org/content/10.1101/2020.04.09.033308v2
Credits
Licence
GPL-3